X
Код презентации скопируйте его
Мукополисахаридозы
Скачать эту презентацию

")
.")




. Рентгенологические признаки синдрома Гурлер — изменения ребер и поз...")
. Рентгенологические признаки синдрома Гурлер — деформация таза и бед...")
. Рентгенологические признаки синдрома Гурлер — характерный вид кистей.")
.")

.")
. Синдром Гунтера у мальчика 2 лет — грубые черты лица, скафоцефалия.")
. Синдром Гунтера у мальчика 2 лет — изменения скелета слабо выражены...")
.")
.")

. Рентгенологические признаки синдрома Моркио — искривление позвоночн...")
. Рентгенологические признаки синдрома Моркио — деформация костей таз...")
. Рентгенологические признаки синдрома Моркио — деформация костей кисти.")
")
. Синдром Марото — Лами: (типичные внешние проявления у девочки 9 лет...")
. Синдром Марото — Лами: (типичные внешние проявления у девочки 9 лет...")
. Рентгенологические признаки синдрома Марото — Лами — изменения позв...")
. Рентгенологические признаки синдрома Марото — Лами — деформация кос...")



Презентация на тему Мукополисахаридозы
Скачать эту презентациюCлайд 1
 Мукополисахаридозы Подготовила Агеева А.В.
Мукополисахаридозы Подготовила Агеева А.В.
Cлайд 2
") Мукополисахаридозы (мукополисахариды + -ōsis)
Мукополисахаридозы (мукополисахариды + -ōsis)
Cлайд 3
.") Мукополисахаридоз типа I-Н (синдром Гурлер).
Мукополисахаридоз типа I-Н (синдром Гурлер).
Cлайд 4
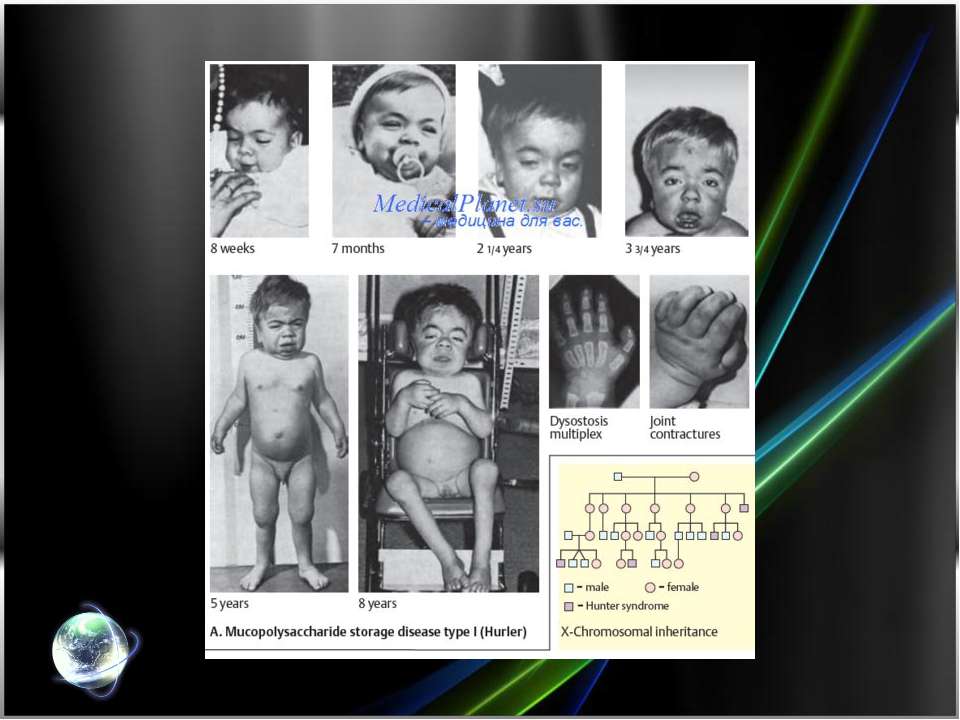
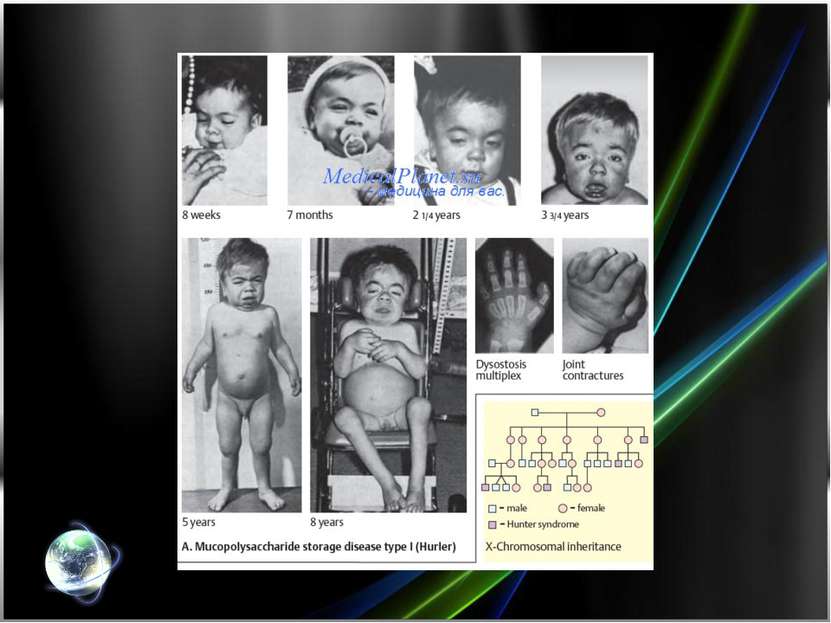
 Рис. 1. Синдром Гурлер: типичные внешние проявления.
Рис. 1. Синдром Гурлер: типичные внешние проявления.
Cлайд 5
 Аномалии лицевого скелета при синдроме Hurler
Аномалии лицевого скелета при синдроме Hurler
Cлайд 6
 Помутнение роговицы при синдроме Hurler Мукополисахаридозы Группа наследственных заболеваний, обусловленных дефицитом ферментов гидролиза мукополисахаридов (глюкозидазы). Продукты незавершенного обмена аккумулируются во внутриклеточных вакуолях в различных тканях и органах, обнаруживаются и в моче.
Помутнение роговицы при синдроме Hurler Мукополисахаридозы Группа наследственных заболеваний, обусловленных дефицитом ферментов гидролиза мукополисахаридов (глюкозидазы). Продукты незавершенного обмена аккумулируются во внутриклеточных вакуолях в различных тканях и органах, обнаруживаются и в моче.
Cлайд 7

Cлайд 8
. Рентгенологические признаки синдрома Гурлер — изменения ребер и поз...") Рис. 2а). Рентгенологические признаки синдрома Гурлер — изменения ребер и позвоночника.
Рис. 2а). Рентгенологические признаки синдрома Гурлер — изменения ребер и позвоночника.
Cлайд 9
. Рентгенологические признаки синдрома Гурлер — деформация таза и бед...") Рис. 2б). Рентгенологические признаки синдрома Гурлер — деформация таза и бедренных костей.
Рис. 2б). Рентгенологические признаки синдрома Гурлер — деформация таза и бедренных костей.
Cлайд 10
. Рентгенологические признаки синдрома Гурлер — характерный вид кистей.") Рис. 2в). Рентгенологические признаки синдрома Гурлер — характерный вид кистей.
Рис. 2в). Рентгенологические признаки синдрома Гурлер — характерный вид кистей.
Cлайд 11
.") Мукополисахаридоз типа I-S (болезнь Шейе; поздний синдром Гурлер).
Мукополисахаридоз типа I-S (болезнь Шейе; поздний синдром Гурлер).
Cлайд 12
 Рис. 4. Болезнь Шейе: типичные внешние проявления
Рис. 4. Болезнь Шейе: типичные внешние проявления
Cлайд 13
.") Мукополисахаридоз типа II (синдром Гунтера).
Мукополисахаридоз типа II (синдром Гунтера).
Cлайд 14
. Синдром Гунтера у мальчика 2 лет — грубые черты лица, скафоцефалия.") Рис. 3а). Синдром Гунтера у мальчика 2 лет — грубые черты лица, скафоцефалия.
Рис. 3а). Синдром Гунтера у мальчика 2 лет — грубые черты лица, скафоцефалия.
Cлайд 15
. Синдром Гунтера у мальчика 2 лет — изменения скелета слабо выражены...") Рис. 3б). Синдром Гунтера у мальчика 2 лет — изменения скелета слабо выражены, нет кифоза, контрактур.
Рис. 3б). Синдром Гунтера у мальчика 2 лет — изменения скелета слабо выражены, нет кифоза, контрактур.
Cлайд 16
.") Мукополисахаридоз типа III (синдром Санфилиппо, болезнь Санфилиппо).
Мукополисахаридоз типа III (синдром Санфилиппо, болезнь Санфилиппо).
Cлайд 17
.") Мукополисахаридоз типа IV (снидром Моркио, болезнь Моркио).
Мукополисахаридоз типа IV (снидром Моркио, болезнь Моркио).
Cлайд 18
 Рис. 5. Синдром Моркио: типичные внешние проявления.
Рис. 5. Синдром Моркио: типичные внешние проявления.
Cлайд 19
. Рентгенологические признаки синдрома Моркио — искривление позвоночн...") Рис. 6а). Рентгенологические признаки синдрома Моркио — искривление позвоночника и платиспондилия.
Рис. 6а). Рентгенологические признаки синдрома Моркио — искривление позвоночника и платиспондилия.
Cлайд 20
. Рентгенологические признаки синдрома Моркио — деформация костей таз...") Рис. 6б). Рентгенологические признаки синдрома Моркио — деформация костей таза, вертлужных впадин, гипоплазия головок бедренных костей.
Рис. 6б). Рентгенологические признаки синдрома Моркио — деформация костей таза, вертлужных впадин, гипоплазия головок бедренных костей.
Cлайд 21
. Рентгенологические признаки синдрома Моркио — деформация костей кисти.") Рис. 6в). Рентгенологические признаки синдрома Моркио — деформация костей кисти.
Рис. 6в). Рентгенологические признаки синдрома Моркио — деформация костей кисти.
Cлайд 22
") Мукополисахаридоз типа VI (синдром Марото — Лами, болезнь Марото — Лами)
Мукополисахаридоз типа VI (синдром Марото — Лами, болезнь Марото — Лами)
Cлайд 23
. Синдром Марото — Лами: (типичные внешние проявления у девочки 9 лет...") Рис. 7а). Синдром Марото — Лами: (типичные внешние проявления у девочки 9 лет) — грубые черты лица, бочкообразная грудная клетка.
Рис. 7а). Синдром Марото — Лами: (типичные внешние проявления у девочки 9 лет) — грубые черты лица, бочкообразная грудная клетка.
Cлайд 24
. Синдром Марото — Лами: (типичные внешние проявления у девочки 9 лет...") Рис. 7б). Синдром Марото — Лами: (типичные внешние проявления у девочки 9 лет) — контрактуры верхних и нижних конечностей.
Рис. 7б). Синдром Марото — Лами: (типичные внешние проявления у девочки 9 лет) — контрактуры верхних и нижних конечностей.
Cлайд 25
. Рентгенологические признаки синдрома Марото — Лами — изменения позв...") Рис. 8а). Рентгенологические признаки синдрома Марото — Лами — изменения позвоночника.
Рис. 8а). Рентгенологические признаки синдрома Марото — Лами — изменения позвоночника.
Cлайд 26
. Рентгенологические признаки синдрома Марото — Лами — деформация кос...") Рис. 8б). Рентгенологические признаки синдрома Марото — Лами — деформация костей таза.
Рис. 8б). Рентгенологические признаки синдрома Марото — Лами — деформация костей таза.
Cлайд 27
 Синдром Моркио-Улльриха. Генетически детерминированное заболевание - мукополисахаридоз - IV. Хондродистрофия характеризуется ослаблением связочного аппарата суставов, значительными деформациями скелета, в частности грудным горбом. Наблюдаются также сердечно-сосудистая недостаточность, деформация ногтей, кривошея. Глазные симптомы: дистрофические изменения, помутнения стромы роговицы, атрофия зрительного нерва, а, б - общий вид ребенка. Инфантильность. Грудная клетка деформирована, искривление грудного отдела позвоночника, кривошея, помутнение роговицы, двусторонний крипторхизм; в - частичная атрофия зрительных нервов; г - помутнение роговицы.
Синдром Моркио-Улльриха. Генетически детерминированное заболевание - мукополисахаридоз - IV. Хондродистрофия характеризуется ослаблением связочного аппарата суставов, значительными деформациями скелета, в частности грудным горбом. Наблюдаются также сердечно-сосудистая недостаточность, деформация ногтей, кривошея. Глазные симптомы: дистрофические изменения, помутнения стромы роговицы, атрофия зрительного нерва, а, б - общий вид ребенка. Инфантильность. Грудная клетка деформирована, искривление грудного отдела позвоночника, кривошея, помутнение роговицы, двусторонний крипторхизм; в - частичная атрофия зрительных нервов; г - помутнение роговицы.
Cлайд 28
 Редко встречаемые типы мукополисахаридозов
Редко встречаемые типы мукополисахаридозов
Cлайд 29
 Диагноз основывается на клинических проявлениях, данных рентгенологического исследования, определении экскреции с мочой гликозаминогликанов, исследовании активности специфических ферментов культуре клеток (фибробластах кожи и лейкоцитах), амниотической жидкости (антенатальная диагностика).
Диагноз основывается на клинических проявлениях, данных рентгенологического исследования, определении экскреции с мочой гликозаминогликанов, исследовании активности специфических ферментов культуре клеток (фибробластах кожи и лейкоцитах), амниотической жидкости (антенатальная диагностика).
Cлайд 30